1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
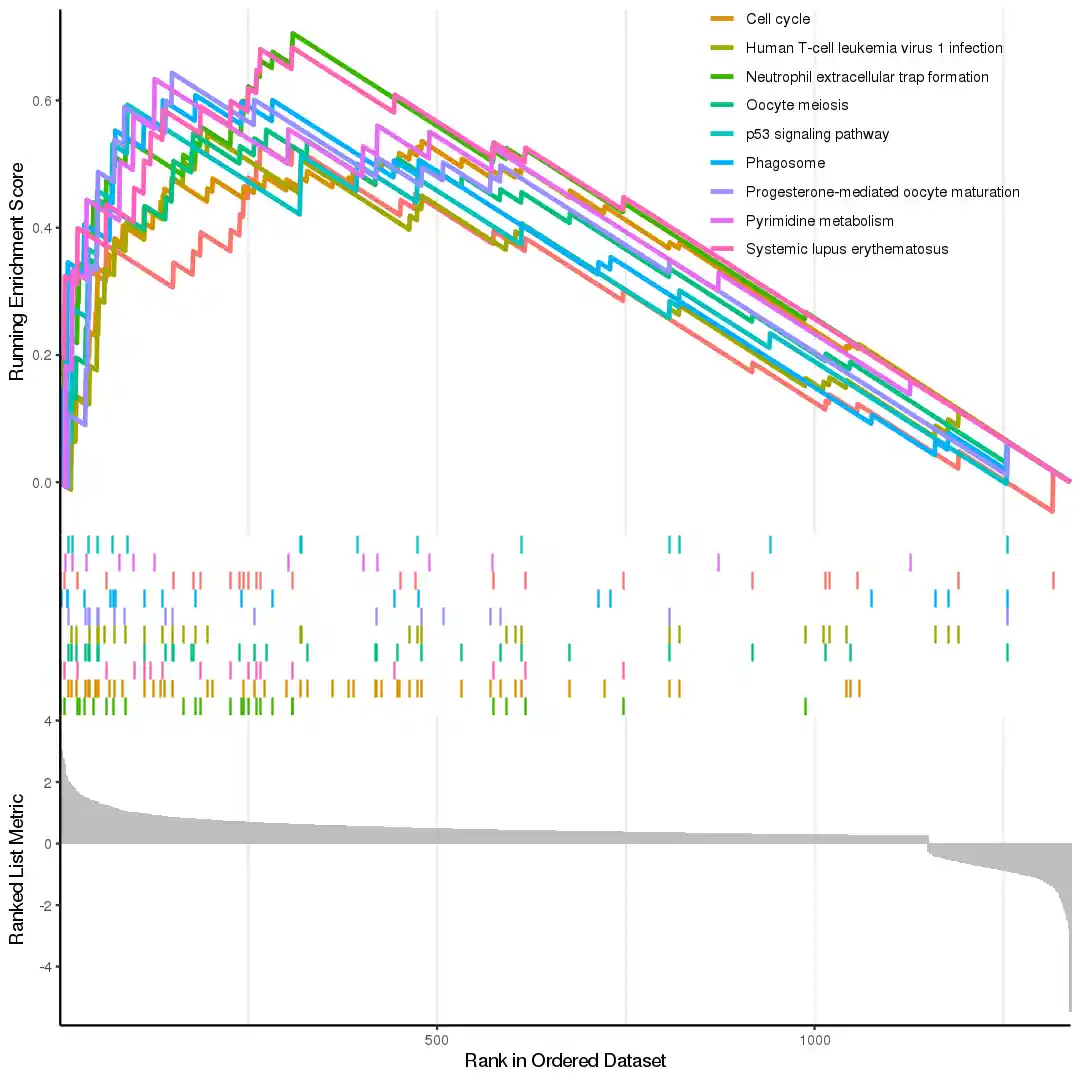
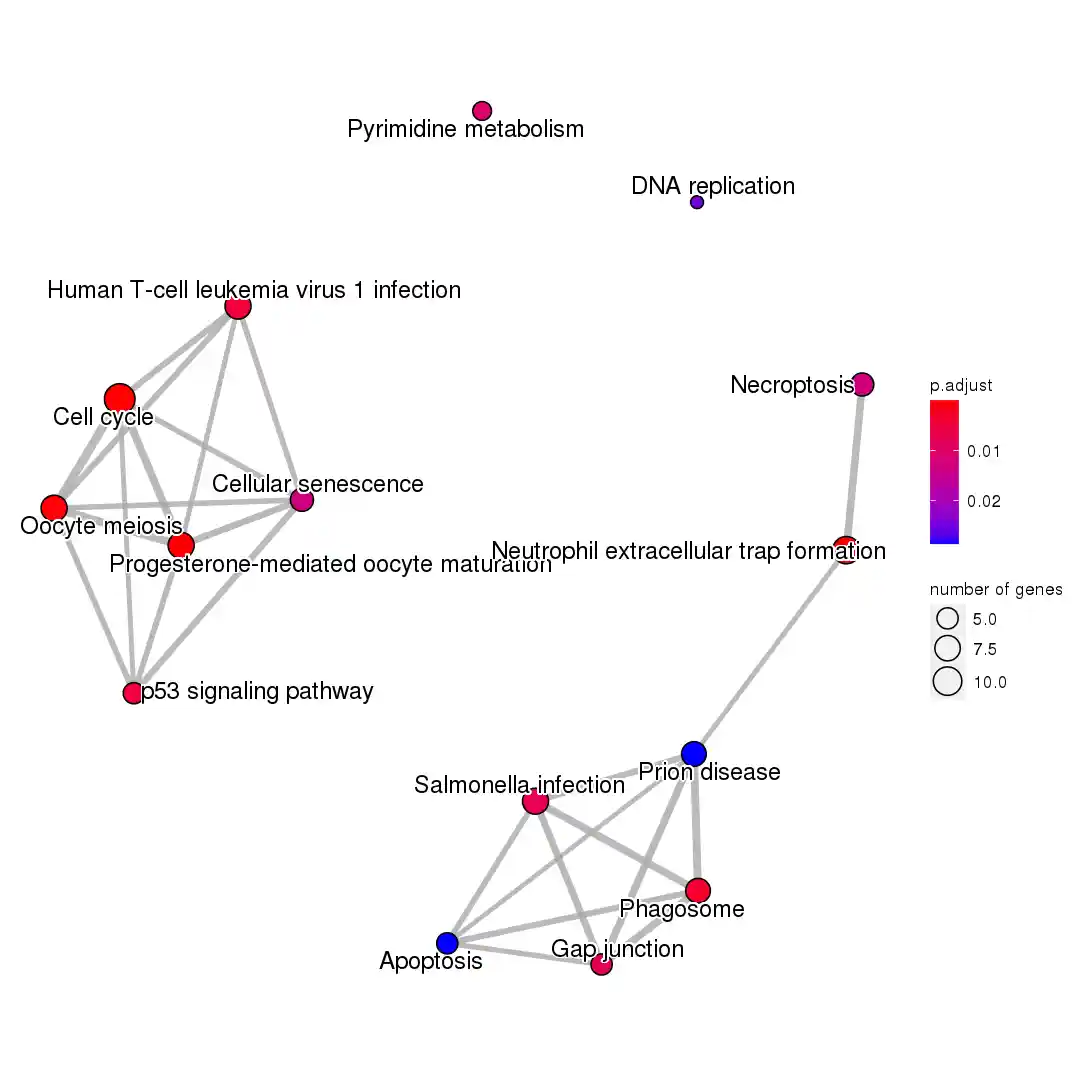
| # clusterProfiler作kegg富集分析
library(clusterProfiler)
library(enrichplot)
library(forcats)
f_id2name_fuck <- function(lc_exp, lc_db){
if(!is.data.frame(lc_db)){
lc_ids <- toTable(lc_db)
}else{
lc_ids <- lc_db
}
res_n <- rownames(lc_exp)
res_n <- res_n[res_n %in% lc_ids[[1]]]
res <- lc_exp[res_n,]
if(!is.data.frame(res)){res=data.frame(row.names = res_n, res)}
lc_ids=lc_ids[match(rownames(res),lc_ids[[1]]),]
lc_tmp = by(res,
lc_ids[[2]],
function(x) rownames(x)[which.max(rowMeans(x))])
lc_probes = as.character(lc_tmp)
res_n <- rownames(res)
res_n <- res_n[res_n %in% lc_probes]
res = res[res_n,]
if(!is.data.frame(res)){res=data.frame(row.names = res_n, res)}
rownames(res)=lc_ids[match(rownames(res),lc_ids[[1]]),2]
res
}
library(org.Hs.eg.db)
f_id2name_sb <- function(lc_cgene, keytype="SYMBOL", columns="ENTREZID"){
res=select(org.Hs.eg.db,keys=lc_cgene,columns=columns, keytype=keytype)
res <- subset(res, !is.na(ENTREZID))
res$ENTREZID
}
library(ggplot2)
library(DOSE)
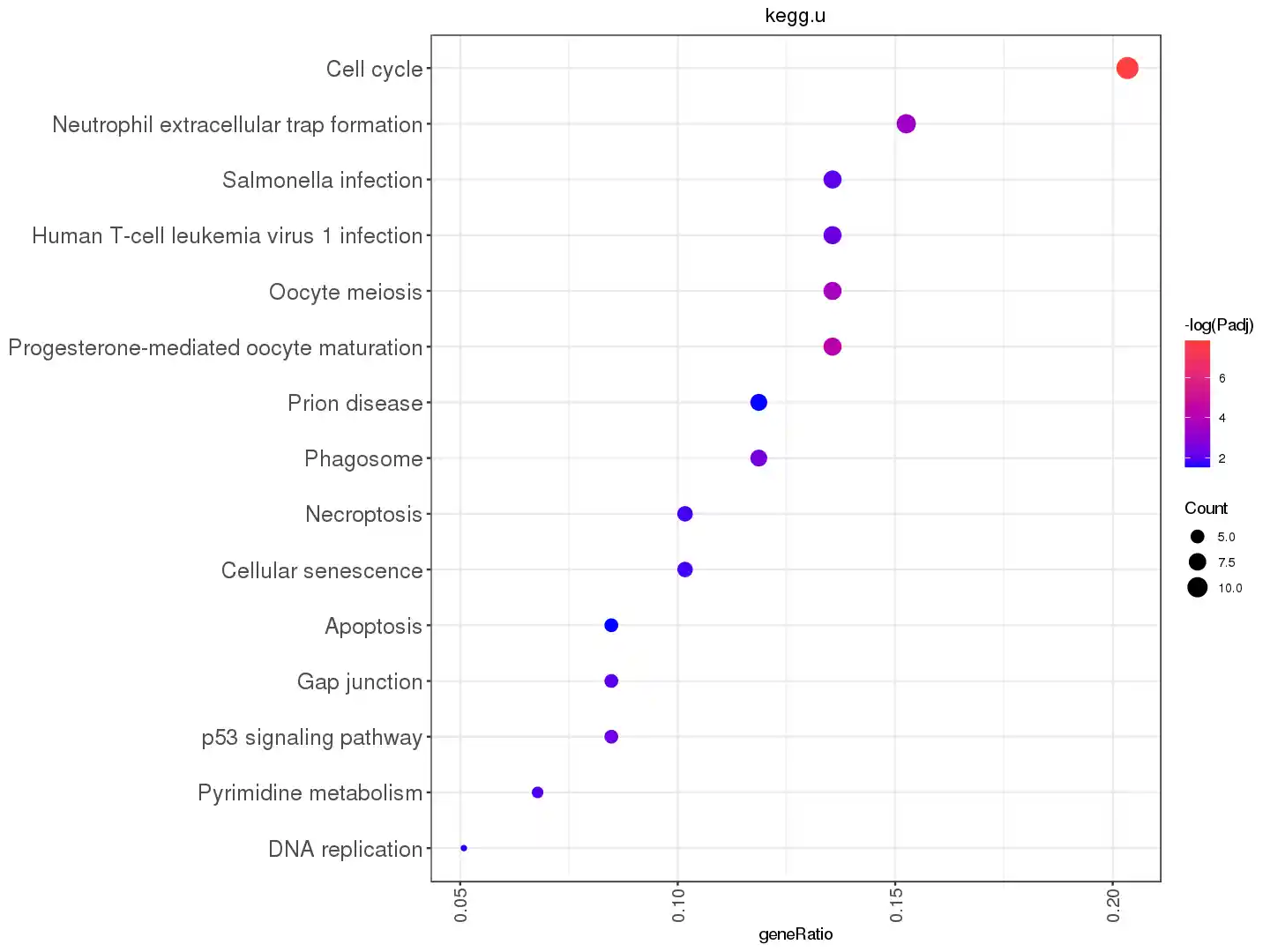
f_kegg_p <- function(keggr2, n = 15){
keggr <- subset(keggr2@result, p.adjust < 0.05)
keggr[['-log(Padj)']] <- -log10(keggr[['p.adjust']])
keggr[['geneRatio']] <- parse_ratio(keggr[['GeneRatio']])
keggr$Description <- factor(keggr$Description,
levels=keggr[order(keggr$geneRatio),]$Description)
ggplot(head(keggr,n),aes(x=geneRatio,y=Description))+
geom_point(aes(color=`-log(Padj)`,
size=`Count`))+
theme_bw()+
scale_color_gradient(low="blue1",high="brown1")+
labs(y=NULL) +
theme(axis.text.x=element_text(angle=90,hjust = 1,vjust=0.5, size = 12),
axis.text.y=element_text(size = 15))
}
|