第一步 准备好数据
内有 name_R1.fq.gz 和 name_R2.fq.gz
大鼠的索引文件
上面的染色体模板命名不规范,使用下面脚本修改
1 2 3 4 5 6 7 # !/bin/bash
规范后的染色体命名
两份注释文件的差别 ,一般使用不带chr的文件
第二步 生成排好序的bam文件
nohup /home/gene/xuchen/gp1.bash > /home/gene/xuchen/1.log 2>&1 &
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 # !/bin/sh # 设置CleanData存放目录 # 设置idx存放目录 # 设置这一步的输出目录 # 内存使用量为 6000M * 6 = 36G # 没用的东西删掉
查看 1.log 确保匹配率高于80%,下图为 95.01%
第三步 生成基因矩阵
nohup /home/gene/xuchen/gp2.bash > /home/gene/xuchen/2.log 2>&1 &
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 # 上一步的输出目录 # 规范命名后的染色体模板数据 # GTF数据,一般使用不带chr的 # 整理样本列表, 空格分隔 # 用来生csv的表头,逗号分隔,与样本列表对应 # 设置输出目录
查看处理进度 tail -f /home/gene/xuchen/2.log
更简单的代码:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 # !/bin/bash # 上一步的输出目录 # 设置这一步的输出目录 # 规范命名后的染色体模板数据 # GTF数据,一般使用不带chr的
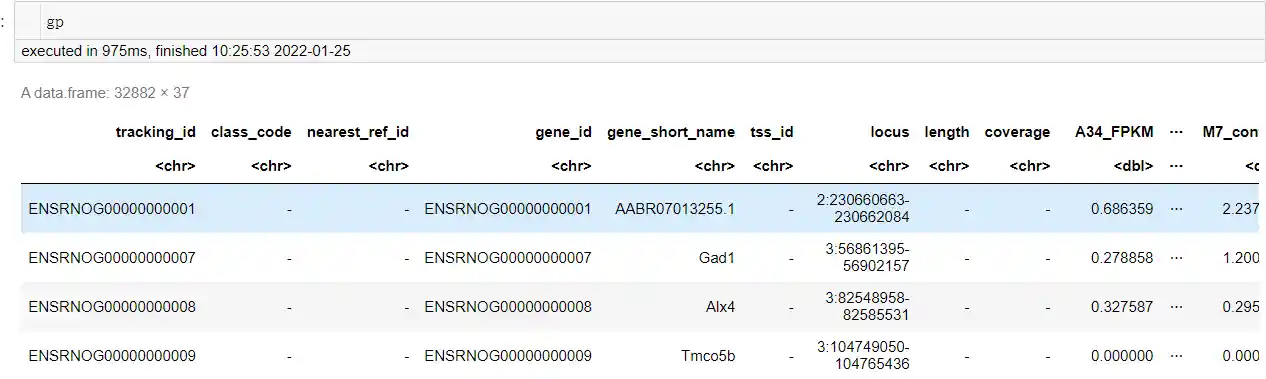
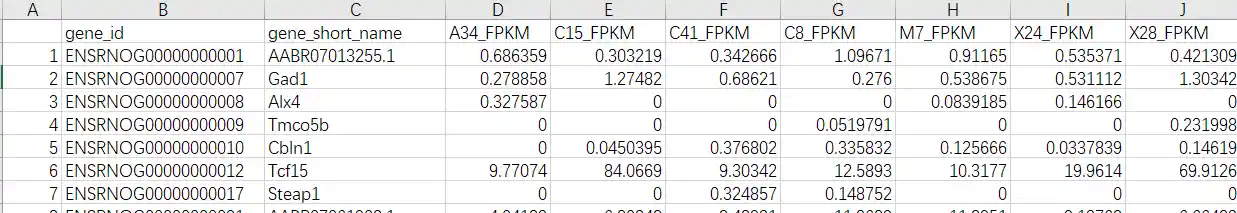
第四步 转换成symbol的csv 1 2 3 4 5 gp <- read.table( '~/gene/xuchen/diffout_gp/genes.fpkm_tracking' , header = T ) <- stringr:: str_detect( colnames( gp) , 'FPKM' ) <- colnames( gp) [ sampleN] <- gp[ , c ( 'gene_id' , 'gene_short_name' , sampleN) ] ( gp_filter, file= 'gp.csv' )
gp的内容
gp.csv的内容