clusterProfiler:自定义数据库
clusterProfiler中提供了enricher和GSEA两个函数,enricher包装了**Over-representation analysis**,GSEA包装了Gene Set Enrichment Analysis。这两个函数与其他的enrichKEGG、gseKEGG的唯一不同点是提供的不是KEGG数据库,而是TERM2GENE和TERM2NAME两个参数。
从gson格式初探中,我们可以得到一个Anno对象kk,恰好其中就有kk@gsid2gene、kk@gsid2name两个属性,因此我们可以得知gsid2gene、gsid2name都是一个两列的长数据框:gsid2gene的第一列是gsid、表示通路ID,第二列是gene、表示通路基因;gsid2name的第一列是gsid,第二列是name、对通路的描述。有了这些知识,我们可以尝试自己构建一个数据库。
构建尝试
包自带的KEGG数据分析起来需要输入ENTREZID。而常规的GTF文件中并不包含这个,通过对比得到的结果只有SYMBOL和ENSEMBL,因此直接转换难免出现不能对应的情况。所以我们需要一个以SYMBOL为gene的gsid2gene数据库。
1 | |
通过上面的代码,我们得到了一个gene为SYMBOL,其他与KEGG完全一致的新数据库,让我们放入enricher中进行测试,看看是否能行。
1 | |
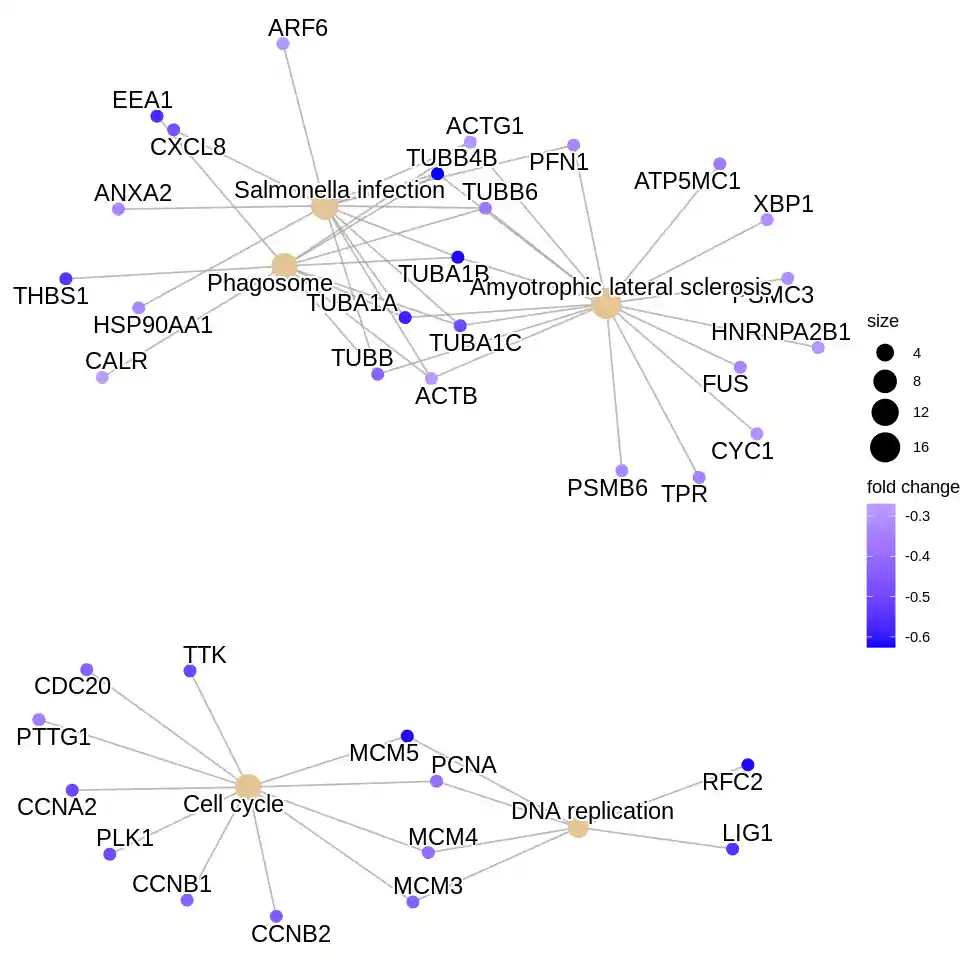
显然,结果是成功的!
豁然开朗
既然可行,那我们来构建HALLMARKS试试。
- 下载 h.all.v7.5.1.symbols.gmt,上传到服务器上
- GMT格式为制表符分隔文件,第一列是通路名称,第二列是描述,第三列以及之后是对应通路下的基因,每个通路为一行。
- 使用下面的代码构建我们自己的clusterProfiler数据库
1 | |
得到自己的HALLMARKS后,使用前面的方法即可进行enricher和GSEA分析。
1 | |
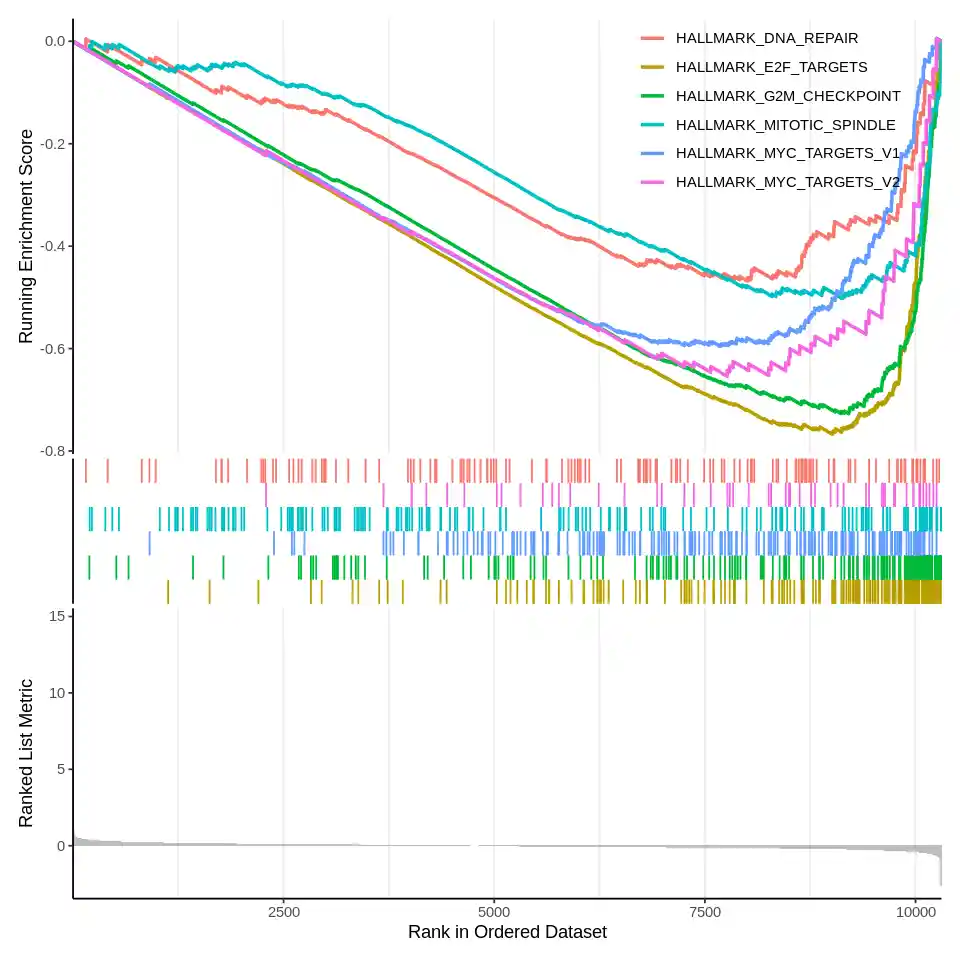
clusterProfiler:自定义数据库
https://b.limour.top/1923.html