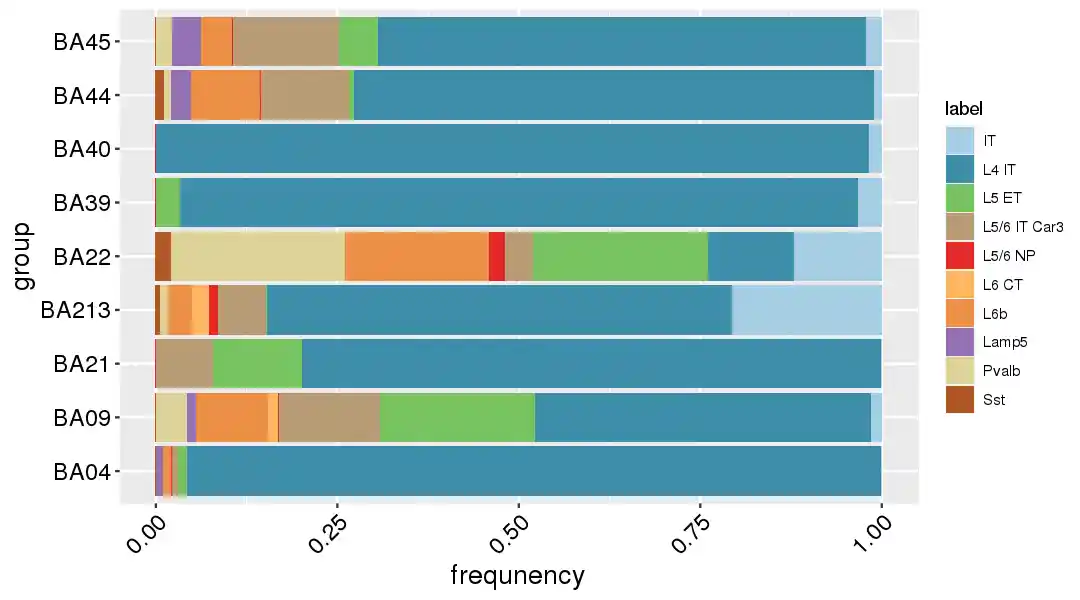
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
| Idents(sc_Neuron) <- sc_Neuron[['hmca_class']]
all_markers <- FindAllMarkers(sc_Neuron, min.pct = 0.25, logfc.threshold = 0.25)
significant_markers <- subset(all_markers, subset = p_val_adj<0.05)
f_DEG_Volcano <- function(lc_logFC, lc_p, lc_gene, Threshold_logFC = 1, Threshold_p = 0.05, lc_rep=1:10){
col_vector = rep(rgb(108, 200, 228, maxColorValue = 255), length(lc_logFC))
col_vector[lc_p < Threshold_p & lc_logFC > Threshold_logFC] = rgb(226, 61, 75, maxColorValue = 255)
col_vector[lc_p < Threshold_p & lc_logFC < -Threshold_logFC] = rgb(232, 168, 71, maxColorValue = 255)
lc_p[lc_p < 1e-10] = 1e-10
lc_p[lc_p > 1 is.na(lc_p)] = 1
df = data.frame(logFC <- lc_logFC, `-log10(P)` <- -log10(lc_p), col <- col_vector, gene <- lc_gene)
colnames(df) <- c('logFC', '-log10(P)', "col", "gene")
lc_tp_logFC <- df$logFC
lc_tp_logFC[lc_p>=Threshold_p] = 0
lc_idx <- order(lc_tp_logFC)[c(lc_rep, length(lc_gene)+1-lc_rep)]
df$logFC[df$logFC > 10] = 10
df$logFC[df$logFC < -10] = -10
res <- ggplot() + geom_point(aes(logFC, `-log10(P)`, col=I(col)), data = df)
res <- res + theme_bw() + theme(panel.grid=element_line(colour=NA))
res <- res + geom_hline(yintercept=-log10(Threshold_p), linetype="longdash")
res <- res + geom_vline(xintercept=c(Threshold_logFC, -Threshold_logFC), linetype="longdash")
res <- res + geom_text_repel(data=df[lc_idx,],aes(logFC,`-log10(P)`,label=gene), force=T, max.overlaps=Inf)
res
}
|